韓國(guó)醫(yī)療器械市場(chǎng)準(zhǔn)入的流程相對(duì)復(fù)雜,涉及到多個(gè)步驟和多個(gè)部門的參與。本文將簡(jiǎn)單介紹一下醫(yī)療器械出口韓國(guó)需要的認(rèn)證和注冊(cè)流程。
主管部門
韓國(guó)食品藥品管理局(MFDS)是韓國(guó)醫(yī)療器械的主管部門,負(fù)責(zé)食品,藥品,醫(yī)療器械和化妝品的安全,食品和制藥工業(yè)發(fā)展,以及促進(jìn)公共健康。
醫(yī)療器械分類
韓國(guó)將醫(yī)療器械分為四類,即Ⅰ、Ⅱ、Ⅲ、Ⅳ類,這種分類方法與歐盟對(duì)醫(yī)療器械的分類方法非常相似。每一類器械都有不同的市場(chǎng)準(zhǔn)入途徑,基于醫(yī)療器械的風(fēng)險(xiǎn)分類,審批流程和時(shí)間也有所不同。
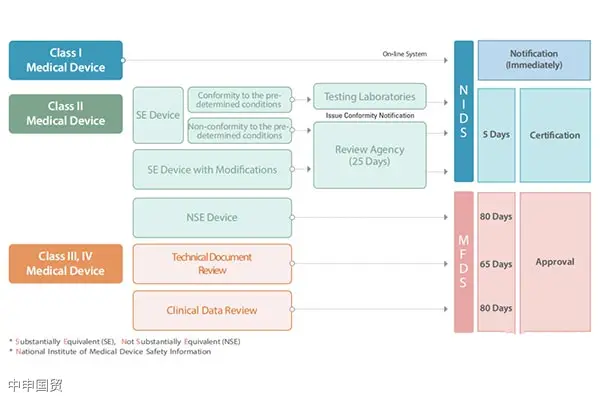
產(chǎn)品測(cè)試與臨床試驗(yàn)
需要注冊(cè)的產(chǎn)品樣品可直接寄到韓國(guó)實(shí)驗(yàn)室測(cè)試,或在具有韓國(guó)相應(yīng)資質(zhì)的國(guó)內(nèi)實(shí)驗(yàn)室進(jìn)行測(cè)試,提供合格的測(cè)試報(bào)告。MFDS指定了一系列的測(cè)試實(shí)驗(yàn)室和臨床試驗(yàn)的定點(diǎn)醫(yī)院。
體系考核
出口到韓國(guó)的II,III,IV類醫(yī)療器械制造商都需要符合KGMP(Korean Good Manufacturing Practice)的要求,KGMP的要求與ISO 13485相類似。KGMP證書是頒發(fā)給進(jìn)口商而不是生產(chǎn)商,證書每3年更新一次。
韓國(guó)化妝品證書申請(qǐng)網(wǎng)站:https://kcia.or.kr/cert/main/
韓國(guó)醫(yī)療器械注冊(cè)流程
韓國(guó)進(jìn)口醫(yī)療器械分為以下幾個(gè)步驟:選擇韓國(guó)持證人,產(chǎn)品注冊(cè),資料準(zhǔn)備,產(chǎn)品檢測(cè),臨床試驗(yàn),產(chǎn)品注冊(cè)證書,KGMP資料準(zhǔn)備,醫(yī)院準(zhǔn)入。
產(chǎn)品上市以及上市后的監(jiān)管
產(chǎn)品注冊(cè)證以及KGMP證書頒發(fā)后,對(duì)于非家用醫(yī)療器械,還需要做醫(yī)院的準(zhǔn)入,進(jìn)入醫(yī)院的醫(yī)保系統(tǒng),取得醫(yī)院醫(yī)保號(hào)。此后,產(chǎn)品就可以正式在韓國(guó)市場(chǎng)銷售了。產(chǎn)品上市后,MFDS有權(quán)跟蹤一些指定的高風(fēng)險(xiǎn)醫(yī)療器械產(chǎn)品。對(duì)于在韓國(guó)市場(chǎng)出現(xiàn)不良反應(yīng)的產(chǎn)品應(yīng)及時(shí)召回。
最后,國(guó)外產(chǎn)品要進(jìn)入韓國(guó)市場(chǎng)的條件有:需要準(zhǔn)備非常詳細(xì)的產(chǎn)品技術(shù)文檔和申請(qǐng)韓國(guó)KGMP需要的資料;公司的體系要符合ISO 13485的要求;產(chǎn)品要符合標(biāo)準(zhǔn)要求。以上就是韓國(guó)醫(yī)療器械準(zhǔn)入的相關(guān)制度,希望對(duì)醫(yī)療器械的進(jìn)出口企業(yè)有所幫助。
標(biāo)簽: 出口韓國(guó) · 醫(yī)療器械進(jìn)出口



? 2025. All Rights Reserved. 滬ICP備2023007705號(hào)-2  滬公網(wǎng)安備31011502009912號(hào)
滬公網(wǎng)安備31011502009912號(hào)