Home ? Medical Equipment ? Certification Requirements and Procedures for Exporting Medical Devices to Australia
Australia has a mature and large - scale public and private healthcare system, and there is also a strong demand forMedical EquipmentHowever, to sell, import or export medical devices in Australia, it is necessary to complete the registration review and obtain the access certificate in accordance with relevant regulations. This article will introduce this process in detail.
I. Australian Regulatory Regulations
As early as 1966, Australia passed the Therapeutic Goods Act to regulate therapeutic goods. The main current management acts are the Therapeutic Goods Act of 1989 and the Medical Devices Regulations promulgated in 2002. The implementation and supervision of these two regulations are the responsibility of the Therapeutic Goods Administration (TGA), a subordinate agency of the Australian Federal Department of Health and Aged Care. Therefore, manufacturers who want to sellimport and exportmedical devices to Australia need to submit a market access application to the TGA.
II. Definition and Classification of Medical Devices in Australia
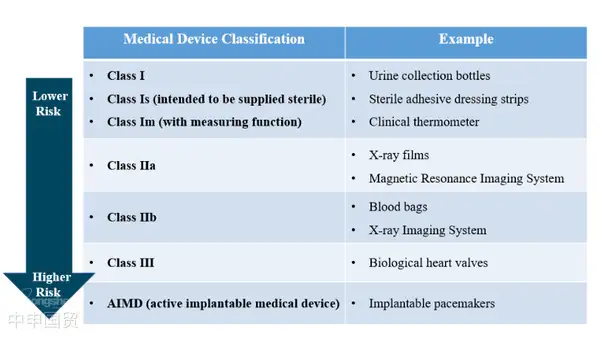
Australia defines medical devices as therapeutic goods consisting of tools, instruments, appliances or other items, as well as necessary accessories or software. These therapeutic goods mainly achieve their primary effects through non - pharmacological, immunological or metabolic means, although these means may assist their effects. According to the risk level, medical devices are classified into Class I, Class IIa, Class IIb, Class III, and AIMD (Active Implantable Medical Devices).
III. Market Access of Medical Devices in Australia
The TGA manages medical devices in three categories: exemption, filing and registration. Each type of medical device must obtain government approval before being put on the market and meet the basic requirements of medical devices. High - risk medical devices must pass the TGAs assessment and obtain approval before being launched. Low - risk medical devices can be self - assessed by enterprises and can enter the market as long as they meet the quality and safety conditions. All medical devices produced in Australia must meet the GMP standards and be produced in a clean and pollution - free environment.
IV. Analysis of the Market Launch Process of Medical Devices in Australia
The market launch process of medical devices in Australia is a rigorous and cumbersome task, which requires fulfilling the responsibilities of multiple key roles, clarifying the definition and risk level of medical devices, and completing the conformity assessment and ARTG registration.
(1) Clarify Key Roles
First of all, the registration and market launch process of medical devices in Australia involves two key roles, namely the manufacturer and the sponsor. The manufacturer is responsible for the design, production, packaging, shipping, etc. of the medical device, while the sponsor is an Australian citizen or company who is responsible for applying for a Sponsor registration number from the TGA (Therapeutic Goods Administration) to help complete the registration process.
(2) Clarify the Definition and Risk Level of Medical Devices
Next, it is necessary to clarify the definition and risk level of medical devices. The Therapeutic Goods Act of 1989 defines medical devices and their key points as follows:

In Australia, medical devices that meet the definition of the Therapeutic Goods Act 1989 need to submit a registration application to the TGA. According to different risk levels and intended uses, medical devices are divided into five risk levels. The higher the risk level, the stricter the TGAs supervision.
(3) Apply for a Conformity Assessment Certificate
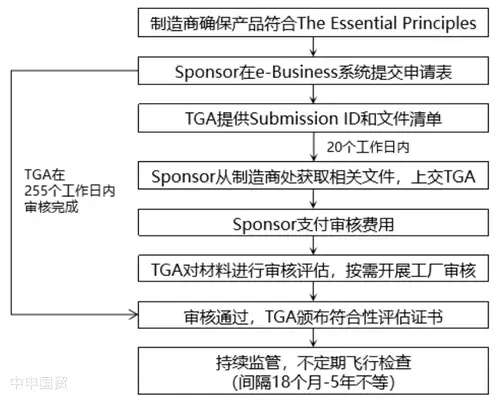
Before medical device registration and market entry, manufacturers must implement qualified conformity assessment procedures, undergo TGA audits, and obtain valid conformity assessment certificates. TGA-recognized conformity assessment certificates include TGA Conformity Assessment Evidence, EC Certificate, and certificates issued based on EC-MRA and EFTA-MRA.
If choosing TGA conformity assessment evidence, the certificate application process is as follows:
(4) Submit ARTG Registration
After completing the conformity assessment, the manufacturer and the Sponsor need to submit ARTG registration to obtain an access certificate for a single medical device to complete the product listing. Only when a medical device is listed in the Australian Register of Therapeutic Goods (ARTG) can it be sold on the Australian market.

V. Quality System of Medical Devices in Australia
The government stipulates that the production processes of all medical device manufacturers must meet the quality requirements related to the devices they produce and have means and procedures for quality assurance. To achieve this goal, Australia has implemented the Good Manufacturing Practice (GMP) system and also moved closer to the ISO 9000 quality system standards. Since 2017, Australia has started to implement the Medical Device Single Audit Program and accepts MDSAP certification certificates provided by qualified inspection institutions.
VI. Post - market Management of Medical Device Products in Australia
This is mainly achieved through the post - market vigilance management system. Australia ensures that all post - market medical devices comply with regulations through the investigation reports of adverse events, laboratory tests and monitoring activities of listed products. Its regulations on post - market adverse event monitoring are very mature, with detailed procedures, combining principles and flexibility, and having strong operability.
VII. Regulations on Registration Changes of Medical Device Products in Australia
After a product obtains access to the market, its information and status still need to be continuously supervised. If there are substantial changes to the product, the manufacturer and the Sponsor must promptly notify the Therapeutic Goods Administration (TGA). The Sponsor needs to log in to the e - Business system to submit a registration change application. These changes may include changes in the manufacturer, sponsor, product information, conformity assessment certificate information, intended use of the product, clinical indications, as well as changes in the product category and GMDN code.
In general, to sell, import or export medical devices in Australia, it is necessary to go through a series of strict access and registration processes. These processes ensure the quality, safety and effectiveness of medical devices, and thus guarantee the quality of the healthcare system.
Tags: Export to Australia Foreign Trade Documents Import and export of medical devices
Related Recommendations





? 2025. All Rights Reserved. Shanghai ICP No. 2023007705-2  PSB Record: Shanghai No.31011502009912
PSB Record: Shanghai No.31011502009912